Aβ42 oligomers trigger synaptic loss through CAMKK2-AMPK-dependent effectors coordinating mitochondrial fission and mitophagy
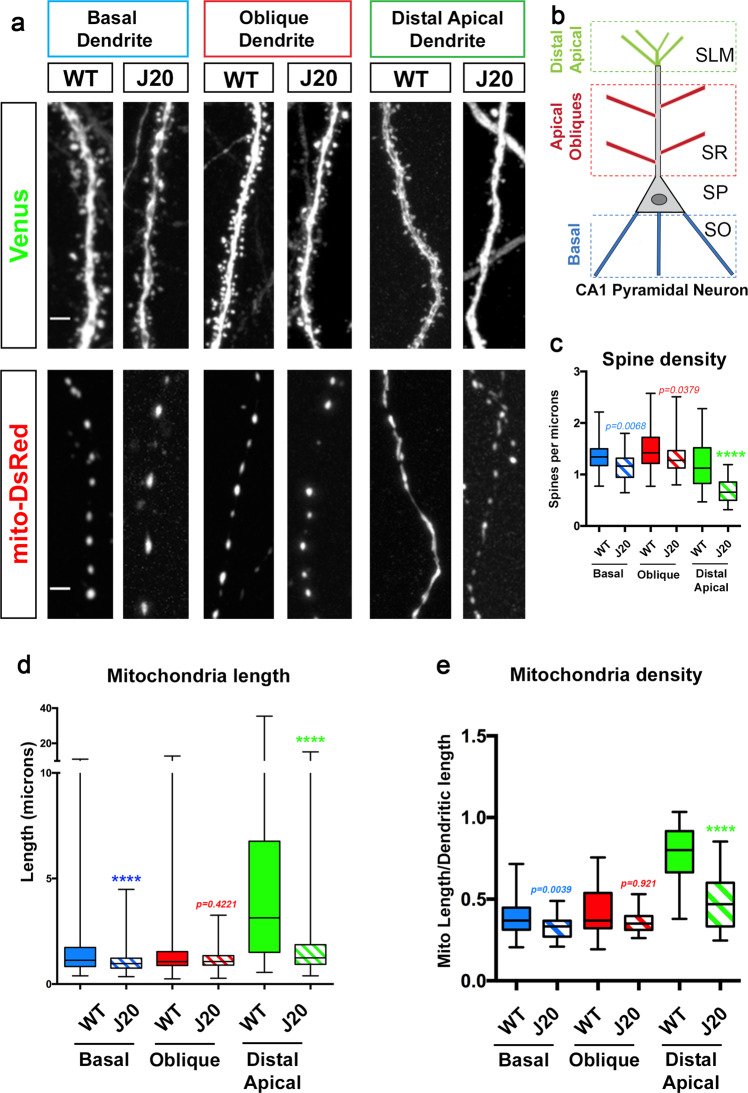
During the early stages of Alzheimer's disease (AD) in both mouse models and human patients, soluble forms of Amyloid-β 1-42 oligomers (Aβ42o) trigger loss of excitatory synapses (synaptotoxicity) in cortical and hippocampal pyramidal neurons (PNs) prior to the formation of insoluble amyloid plaques. In a transgenic AD mouse model, we observed a spatially restricted structural remodeling of mitochondria in the apical tufts of CA1 PNs dendrites corresponding to the dendritic domain where the earliest synaptic loss is detected in vivo. We also observed AMPK over-activation as well as increased fragmentation and loss of mitochondrial biomass in Ngn2-induced neurons derived from a new APPSwe/Swe knockin human ES cell line. We demonstrate that Aβ42o-dependent over-activation of the CAMKK2-AMPK kinase dyad mediates synaptic loss through coordinated phosphorylation of MFF-dependent mitochondrial fission and ULK2-dependent mitophagy. Our results uncover a unifying stress-response pathway causally linking Aβ42o-dependent structural remodeling of dendritic mitochondria to synaptic loss.